
Poster
Raising the bar in preclinical oncology
New oncology drugs only have a 3.4% success rate once making it from Phase I clinical trials to FDA approval. This is the lowest success rate among the 21 major disease indications.1 This poor success rate leaves everyone, from the patients to the benchtop scientists, questioning the validity of the models and the resulting data. This is nothing new in the world of cancer research; we are all attempting to treat a disease that has the ability to fool its host and adapt to survive.
Probability (%) of success(2) by clinical trial phase and therapeutic area
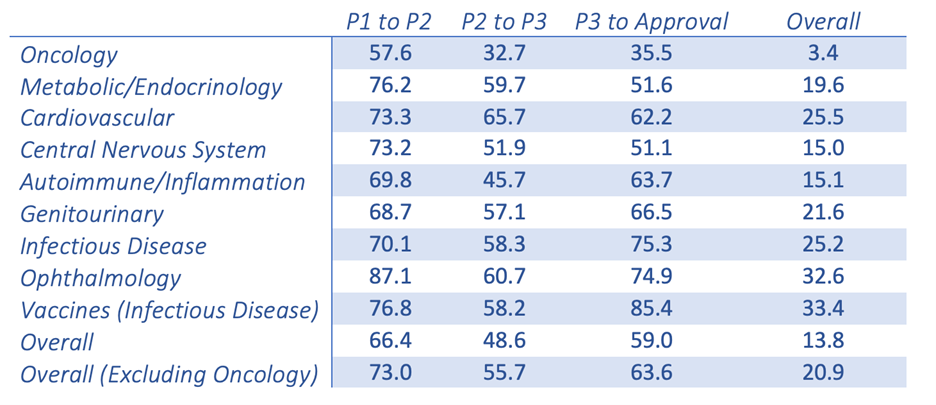
Chi Heem Wong, Kien Wei Siah, Andrew W Lo. Estimation of clinical trial success rates and related parameters. Biostatistics 2019;20(2):273?286.
Adapted from the Wong, Siah and Lo publication (table shown above), it is clear that there is a fundamental disconnect between preclinical data and clinical results. Nine out of 10 attempts to bring a new oncology therapy to the clinic will fail. This is far less than other therapeutic areas, such as cardiovascular and inflammation. The very low success rate in oncology leads drug companies and the FDA to be more lenient when it comes to providing new therapies to the desperate patient population. Unfortunately, this has resulted in drugs that quickly fail in the clinical setting. In a span of ~10 years, there were 9,985 new drugs entering Phase I clinical trials with 31.2% (3,163) for oncology indications alone.1,2 The high failure rate leads to industry criticism that the traditional preclinical animal models are, at best, limited in their power of predictability and, at worst, grossly inaccurate. This may be the easy scapegoat. Improving and evolving the current preclinical animal models is essential to understanding their potential and, equally as important, their limitations. Traditional preclinical (xenograft and syngeneic) models have worked in the past and continue to be highly valuable tools. However, the interpretation of the data that are produced by these models and how they are used to predict clinical response may be where the biggest discrepancies lie.
To mimic more closely the human disease state, many human and murine tumor cell lines have been further validated as orthotopic implants. Implanting the human or murine tumor cells in the tissue of origin can result in a pathological profile that recapitulates human disease and can increase the rate of metastatic involvement when compared to the traditional subcutaneous models.3 As with all models, there are limitations with orthotopic implants; primarily, monitoring disease progression can be limited to survival endpoints which that are not ideal. The optimal situation is to take advantage of the orthotopic implant by using either clinically translatable imaging technology (Magnetic resonance imaging or computerized tomography) or optical imaging (Bioluminescence imaging or fluorescence molecular tomographic imaging). The ability to use imaging technology (clinically translatable or optical) allows for evaluation of a solid tumor or hematological cancer in the same animal over time, as the work is done in the clinic. Tracking disease burden and response in this manner has the potential to be a powerful tool in translating preclinical activity (response) of a new drug into clinical success.
In recent years, there have been significant enhancements in the utilization of patient-derived xenografts (PDXs). In a retrospective analysis of cytotoxic and targeted therapies, PDX models were clearly predictive (~90% accurate) of clinical outcome when dosed at clinically relevant dosage levels.3 This is a significant improvement in the cancer research field, but there are limitations with these models as well. Obtaining fresh human tissue is challenging and chances of successful engraftment, even in a severely immunocompromised mouse, is approximately 30%.3 If engraftment is successful, maintaining these PDX lines as low passage models for future use presents even more complications. The logistics of obtaining and maintaining models, from tissue acquisition to running efficacy studies, along with the overall cost, may be the most significant hindrances to the widespread use and acceptance of these PDX models.
Clinically, the most frequently utilized endpoints to evaluate the effectiveness of a therapy are an industry-standardized set of terms and definitions (Response Evaluation Criteria in Solid Tumors or RECIST criteria). Paramount to this list is the responsiveness of the disease to treatment: complete response (CR), partial response (PR), and overall increase in survival. CRs are defined as complete regressions of the primary tumor mass. PRs are defined as a partial reduction in the primary tumor by ~30%. Therapies are considered successful if they induce either CRs or PRs that can lead to a positive impact on survival. However, in the preclinical setting the commonly used endpoints are tumor-growth inhibition and tumor-growth delay; both are defined as a slowing of disease progression. Unfortunately, tumor-growth inhibition does not directly correlate to an overall increase in survival. This is a critical difference; the evaluation of preclinical efficacy data within the research community is less rigorous and held to a different set of standards than its clinical counterparts. The lower standards allow for more drugs to go through to clinical trials resulting in a greater number of clinical failures. This idea of aligning our preclinical and clinical standards is not novel. In an editorial published in the Journal of the National Cancer Institute, the authors called for a consensus among drug developers that unequivocally defines successful preclinical endpoints.4 If we can collectively raise the bar to more stringent preclinical criteria for the evaluation of novel cancer treatments, we could possibly reduce the failure rate in the clinic. Downstream of this, it would then boost the confidence of clinicians and patients when they are reviewing preclinical data.
Now we all know that change is hard and expecting anything to happen overnight is naïve, but small changes in perceptions and practices now could have significant impact later. The preclinical models that we have invested years and years in developing are effective, if we use them correctly. This starts with concise protocol design and ends with consistent data evaluations. In clinical trials, new drugs will be facing patients with established disease. The research community can use this assumption to design more rigorous preclinical experiments. In the clinic, tumors are well established within the origin tissue with a vascular bed in place to ensure survival at the time treatment is initiated. We can mimic this environment preclinically with either subcutaneous or orthotopic implants by allowing the tumor to grow and become more established. The time that it takes for tumors to become established is highly dependent on the tumor line and the implant location. Subcutaneous tumors can be monitored easily with standard calipers to ensure progressive growth. It is trickier with orthotopic models to ensure that the tumor is actively growing unless you have the ability to image the tumors over time. This relatively straightforward step would save the time and money that is wasted on false positive results generated from studies designed to treat tumors that are not at all or only barely established. At the time of final data analyses, scientists and drug developers need to alter how they define an active new drug. Using the clinical standards as a guideline for activity would decrease the number of new drugs that inevitably fail being pushed into the clinic. This starts with a shift away from tumor-growth inhibition endpoints to the clinically translatable endpoints CRs, PRs, and overall increase in survival.
This is no small request. Trying to convince an entire field of scientists and drug developers to look at their preclinical efficacy data more diligently and to hold it to a more robust set of standards will clearly impact the perceived success rate. However, if more effort is invested into optimizing these new drugs preclinically rather than pushing them into the clinic too early, their chances of real success would increase. All of this, in the long run, would save time, money, and lives potentially.
For more information regarding Labcorp?s Preclinical tumor models and imaging capabilities, please visits: https://drugdevelopment.labcorp.com/industry-solutions/oncology/preclinical/models.html or
References
1 Chi Heem Wong, Kien Wei Siah, Andrew W Lo. Estimation of clinical trial success rates and related parameters. Biostatistics 2019;20(2):273?286.
2 Kamb A. What?s wrong with our cancer models? Nat Rev Drug Discov. 2005; 4(2):161?165.
3 Ruggeri BA, Camp F, Miknyoczki S. Animal models of disease: Pre-clinical animal models of cancer and their applications and utility in drug discovery. Biochem Pharmacol. 2014; Vol. 87(1):150?161.
4 Bertotti A, Trusolino L. From bench to bedside: does preclinical practice in translational oncology need some rebuilding? J Natl Cancer Inst. 2013;105(19):1426?1427.